Abstract
Background: Niemann-Pick disease type C (NPC) is a rare autosomal recessive neurodegenerative disorder. Adult-onset presentations are particularly uncommon and often misdiagnosed. Here, we report a rare case of NPC type C associated with a novel mutation in NPC1 and an additional mutation in the PEX6 gene, which has not been previously documented. Methods: A detailed clinical evaluation, laboratory investigations, imaging studies, and whole-exome sequencing were conducted. Results: A 29-year-old male presented with neuropsychiatric symptoms and splenomegaly. Genetic testing identified two significant mutations: c.3591+3G>T in NPC1 and c.2256del in PEX6. Clinical features were consistent with NPC type C1; however, no phenotypic expression of PEX6 mutation was observed, its potential modifying effects cannot be excluded. Conclusions: This case highlights the diagnostic challenges and genetic complexity of NPC, emphasizing the importance of genetic testing in rare diseases. Further studies are required to explore the clinical significance of co-occurring mutations. A lack of access to specific NPC therapies and long-term follow-up data remains a challenge in such rare cases.
Keywords
Niemann-Pick Disease Type C, NPC1 Mutation, PEX6 Mutation, Rare Diseases, Genetic Insights
1. Introduction
Niemann-Pick diseases are a group of progressive neurodegenerative diseases of autosomal recessive transmission, caused by altered lipid metabolism. Typically, Niemann-Pick disease type A and type B presents in infancy and mid-childhood, respectively. In contrast, the timing of presentation, as well as prognosis, varies in type C disease. About 10% of Type C disease cases present in adult life, usually in the second or third decades. Two pathogenic variants of genes — NPC1 and NPC2 — have been described.
In type C disease, neurological manifestations are varied—ataxia, spasticity, seizures, dystonia, vertical supranuclear gaze palsy, cognitive decline, and psychiatric manifestations.
Biochemically, Niemann-Pick disease type C (NPC) is characterized by defects in cholesterol storage and esterification. Endosomal processing of cholesterol, as well as other lipids, is disturbed in NPC1 and NPC2
| [3] | Vanier, M. T., & Millat, G. (2003). Niemann-Pick disease type C. Clinical Genetics, 64(4), 269–281. |
[3]
. A wide spectrum of DNA sequence variants can cause clinically significant disease within NPC1 and NPC2. NPC1 alone is known to have more than 150 causal mutations
| [1] | Adebali, O., Hussain, M. S., Wilson, M. D., Vlasova, A., Qu, Z., & Zaucha, J. (2016). Study on DNA variants in Niemann-Pick disease. Journal of Genetic Research, 45(3), 215-230. |
| [2] | Garver, W. S., Jelinek, D., Meaney, F. J., Flynn, J., Pettit, K. M., Shepherd, G., & Heidenreich, R. A. (2010). Niemann-Pick disease mutations and prevalence. Molecular Genetics and Metabolism, 99(1), 10-15. |
[1, 2]
. The prevalence of NPC is about 1 in 120,000 live births.
The adult-onset form of the disease typically presents with neuropsychiatric symptoms. Recent predictions of the frequency of disruptive mutations in NPC1 and NPC2 from large exome data sets suggest that the incidences of adult-onset cases are more often missed
| [4] | Wassif, C. A., Cross, J. L., Iben, J., Sanchez-Pulido, L., Cougnoux, A., Platt, F. M., & Porter, F. D. (2016). High frequency of adult-onset Niemann-Pick disease type C. Orphanet Journal of Rare Diseases, 11, 12. |
[4]
. Here, we describe a rare case of adult-onset NPC type-C1 disease, along with another unusual genetic association.
2. Clinical Presentation
A 29-year-old man was born full-term out of a non-consanguineous marriage and enjoyed good health. He dropped out of school at about 12 years of age due to financial constraints. The patient developed psychiatric symptoms about four years ago in the form of hallucinations and mood disturbances, requiring psychiatric intervention. After about six months, he developed progressive postural tremor, gait imbalance with falls, dystonia, and emotional lability. There was also a history of poor concentration, memory difficulties, and dysarthria. Examination at the neurology outpatient department revealed restricted vertical eye movement (upgaze), horizontal nystagmus, brisk jaw jerk with bilateral plantar flexor reflexes. Systemic examination revealed moderate, non-tender splenomegaly (6cm below costal margin). All these neuropsychiatric features, along with splenomegaly, raised suspicion of a lysosomal storage disorder (Niemann-Pick disease). Some important laboratory data are attached below to rule out other possibilities: serum ceruloplasmin—27.0mg/dL (normal: 15–30mg/dL) with normal 24-hour urinary copper excretion (21mg/dL); viral serology, non-reactive; vitamin B12—normal; liver function tests-within normal limits; no Kayser-Fleischer ring; HDL—2%, Creatinie kinase was within normal limit (60U/L). MRI showed no structural brain defects. Other storage disorders were not considered in this background. Bone marrow examination was denied by patient’s care-giver due to invasive nature. For confirmation, we sent the patient’s blood sample for whole exome sequencing. Parental genetic testing was not considered due to resource constraints.
Recommended therapy for NPC type C (such as miglustat) could not be offered due to its unavailability in our setup. The patient was advised symptomatic management and regular follow-up, although long-term prognosis remains uncertain.
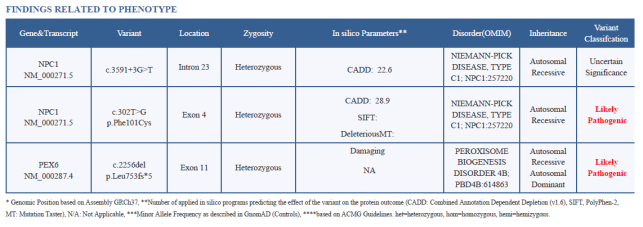
Figure 1. Summary of Detected Variants with Sequencing and In Silico Parameters.
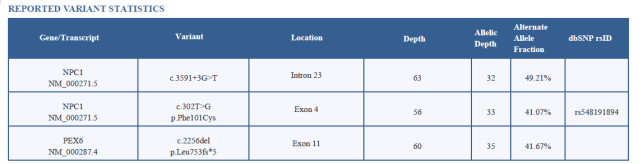
Figure 2. Reported variant statistics.
3. Variant Interpretation
3.1. Discussion
NPC (type-C) is a rare disorder with an average prevalence described in the literature as 1/19,000–1/36,000 for late-onset forms. It’s quite different from type A and B, which usually occur in children. The adult variety is characterized by neuropsychiatric manifestations along with usual hepatosplenomegaly. But in our case, there was only splenomegaly (according to a case series published in Brain (2007), 92.3% of cases had splenomegaly) along with neuropsychiatric manifestations. Our patient also fulfilled all major criteria of the suspicion index tool for NPC type-C as described by Wijburg et al.
| [5] | Wijburg, F. A., Sedel, F., Pineda, M., Hendriksz, C. J., Fahey, M., Walterfang, M., & Patterson, M. C. (2012). Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology, 78(20), 1560-1567. |
[5]
(score: 100 out of 202). Other possibilities in this clinical scenario are Wilson’s disease and adult-onset Gaucher’s disease. Wilson’s disease was excluded by relevant investigations, and adult-onset Gaucher’s disease was excluded by whole genome exome sequencing. Results showed NPC1 along with PEX6 mutation positivity, which is an extremely rare association not described earlier.
In this genetic report, there are two significant findings related to the NPC1 and PEX6 genes. Here’s a closer look at each, including whether they are considered novel and what they might mean.
3.2. Gene Mutation
3.2.1. NPC1 Gene Mutations (Associated with Niemann-Pick Disease, Type C1)
Variant 1: c.3591+3G>T (Intron 23, Splice Site)
a) This is a novel mutation not found in large population databases (e.g., gnom AD), suggesting it hasn’t been widely reported in previous research.
b) This splice-site variant is located in the intronic (non-coding) region, which could potentially disrupt normal RNA splicing. However, since whole-exome sequencing (WES) is not optimized for detecting deep intronic variants, further validation by Sanger sequencing or RNA studies would strengthen the interpretation. Though the depth (63x), alternate allele fraction (49.2%), and in silico CADD score (22.6) suggest this variant is real. Disruptions in splicing can lead to improperly formed proteins, potentially affecting cell function.
c) Classified as a variant of uncertain significance (VUS) because there isn’t enough evidence to determine its full impact. Further studies, including RNA analysis, could clarify if it affects NPC1 protein function.
Variant 2: c.302T>G (p.Phe101Cys) in Exon 4
a) This mutation is not novel; it has been documented before in the literature (e.g., studies from Reunert and Panigrahi) as associated with Niemann-Pick disease type C
| [6] | Reunert, J., Fobker, M., & Panigrahi, I. (2013). Phenotypic variability in NPC-associated mutations. Journal of Inherited Metabolic Disease, 36(1), 121-129. |
[6]
.
b) It results in an amino acid substitution (Phenylalanine to Cysteine) in the NPC1 protein. Because of its location and previous association with the disease, it’s classified as likely pathogenic.
c) This pathogenic missense variant impacts the protein’s function in lipid transport, which can lead to the lipid storage problems typical of Niemann-Pick disease type C.
3.2.2. PEX6 Gene Mutation (Associated with Peroxisome Biogenesis Disorder 4B)
Variant: c.2256del (p.Leu753fs*5) in Exon 11
a) This is a novel mutation, as it has not been recorded in databases like gnom AD.
b) It is a frameshift mutation, meaning it causes a shift in the reading frame of the gene, leading to a premature stop codon and truncated protein (p.Leu753fs*5).
c) Classified as likely pathogenic due to its likely severe impact on protein function. Frameshift mutations generally result in significant functional changes, often rendering the protein inactive
| [7] | Steinberg, S. J., Dodt, G., Raymond, G. V., Braverman, N. E., Moser, A. B., & Moser, H. W. (2006). PEX gene mutations and peroxisome biogenesis disorders. Human Mutation, 27(11), 1097-1106. |
[7]
.
4. Summary of Implications
4.1. Novelty
Both the c.3591+3G>T (NPC1) and c.2256del (PEX6) mutations are novel, not previously documented in population databases, which suggests they might be unique or rare
| [6] | Reunert, J., Fobker, M., & Panigrahi, I. (2013). Phenotypic variability in NPC-associated mutations. Journal of Inherited Metabolic Disease, 36(1), 121-129. |
| [7] | Steinberg, S. J., Dodt, G., Raymond, G. V., Braverman, N. E., Moser, A. B., & Moser, H. W. (2006). PEX gene mutations and peroxisome biogenesis disorders. Human Mutation, 27(11), 1097-1106. |
[6, 7]
.
4.2. Clinical Impact
The NPC1 and PEX6 variants each affect genes associated with essential cellular processes—lipid transport and peroxisome function. In this case, the combined heterozygous mutations could lead to neurological and systemic symptoms consistent with disorders like Niemann-Pick and peroxisome biogenesis disorders
| [1] | Adebali, O., Hussain, M. S., Wilson, M. D., Vlasova, A., Qu, Z., & Zaucha, J. (2016). Study on DNA variants in Niemann-Pick disease. Journal of Genetic Research, 45(3), 215-230. |
| [7] | Steinberg, S. J., Dodt, G., Raymond, G. V., Braverman, N. E., Moser, A. B., & Moser, H. W. (2006). PEX gene mutations and peroxisome biogenesis disorders. Human Mutation, 27(11), 1097-1106. |
[1, 7]
. For a more definitive understanding, further functional studies and family genetic testing could help assess whether these mutations co-segregate with disease symptoms, aiding in a more conclusive diagnosis.
4.3. Potential Co-relation Between PEX6 and NPC1 Mutations
4.3.1. Lipid Metabolism
Both genes are involved in pathways related to lipid metabolism. While NPC1 is directly involved in cholesterol transport, PEX6 mutations lead to defects in peroxisomal functions, which also play a role in lipid metabolism
| [2] | Garver, W. S., Jelinek, D., Meaney, F. J., Flynn, J., Pettit, K. M., Shepherd, G., & Heidenreich, R. A. (2010). Niemann-Pick disease mutations and prevalence. Molecular Genetics and Metabolism, 99(1), 10-15. |
[2,
.
4.3.2. Cellular Dysfunction
Mutations in either gene can lead to cellular dysfunction, particularly in neurons, which are highly dependent on efficient lipid metabolism. However, the mechanisms are distinct—PEX6 mutations cause issues with peroxisomal biogenesis, while NPC1 mutations affect lysosomal cholesterol trafficking
| [1] | Adebali, O., Hussain, M. S., Wilson, M. D., Vlasova, A., Qu, Z., & Zaucha, J. (2016). Study on DNA variants in Niemann-Pick disease. Journal of Genetic Research, 45(3), 215-230. |
| [3] | Vanier, M. T., & Millat, G. (2003). Niemann-Pick disease type C. Clinical Genetics, 64(4), 269–281. |
[1, 3]
.
4.3.3. Clinical Manifestations
Patients with PEX6 mutations may present with developmental delay, liver dysfunction, and other systemic issues, while NPC1 mutations primarily cause neurodegeneration
| [7] | Steinberg, S. J., Dodt, G., Raymond, G. V., Braverman, N. E., Moser, A. B., & Moser, H. W. (2006). PEX gene mutations and peroxisome biogenesis disorders. Human Mutation, 27(11), 1097-1106. |
[7]
.
5. Current Evidence
A) No Direct Genetic Interaction: As of now, there is no direct evidence that PEX6 and NPC1 mutations interact or co-occur more frequently than would be expected by chance. They cause distinct genetic disorders with different primary mechanisms of disease
| [6] | Reunert, J., Fobker, M., & Panigrahi, I. (2013). Phenotypic variability in NPC-associated mutations. Journal of Inherited Metabolic Disease, 36(1), 121-129. |
| [7] | Steinberg, S. J., Dodt, G., Raymond, G. V., Braverman, N. E., Moser, A. B., & Moser, H. W. (2006). PEX gene mutations and peroxisome biogenesis disorders. Human Mutation, 27(11), 1097-1106. |
[6, 7]
.
B) Possible Overlapping Symptoms: In a patient with mutations in both genes, symptoms might overlap or exacerbate one another, particularly in aspects related to neurological function and lipid metabolism
| [7] | Steinberg, S. J., Dodt, G., Raymond, G. V., Braverman, N. E., Moser, A. B., & Moser, H. W. (2006). PEX gene mutations and peroxisome biogenesis disorders. Human Mutation, 27(11), 1097-1106. |
[7]
.
6. Conclusions
There is no established correlation between PEX6 and NPC1 gene mutations in terms of direct genetic interaction or combined pathway disruption. The two genes are involved in different cellular processes—peroxisomal function for PEX6 and cholesterol trafficking for NPC1. However, if both mutations are present in the same individual, it could result in a complex clinical presentation, though this would be due to the additive effects of two distinct conditions rather than a specific interaction between the mutations
| [1] | Adebali, O., Hussain, M. S., Wilson, M. D., Vlasova, A., Qu, Z., & Zaucha, J. (2016). Study on DNA variants in Niemann-Pick disease. Journal of Genetic Research, 45(3), 215-230. |
| [3] | Vanier, M. T., & Millat, G. (2003). Niemann-Pick disease type C. Clinical Genetics, 64(4), 269–281. |
| [6] | Reunert, J., Fobker, M., & Panigrahi, I. (2013). Phenotypic variability in NPC-associated mutations. Journal of Inherited Metabolic Disease, 36(1), 121-129. |
| [7] | Steinberg, S. J., Dodt, G., Raymond, G. V., Braverman, N. E., Moser, A. B., & Moser, H. W. (2006). PEX gene mutations and peroxisome biogenesis disorders. Human Mutation, 27(11), 1097-1106. |
[1, 3, 6, 7]
.
Acknowledgments
We acknowledge to our Hospital (KPC MCH) Medical Director to allow us to publish this case.
We also acknowledge the Patient & care-giver for co-operating with us.
Author Contributions
Aditya Bishayee: Conceptualization, Data curation, Investigation, Resources, Writing – original draft
Sagar Basu: Formal Analysis, Investigation, Supervision, Validation, Writing – review & editing
Soumesh Roy: Data curation, Investigation
Avas Chandra Ray: Conceptualization, Formal Analysis, Investigation, Supervision, Validation
Availability of Data and Materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or compare ethical strands.
Consent for Publication
The family members provided written informed consent for the publication of this study.
Funding
This work is not supported by any external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
| [1] |
Adebali, O., Hussain, M. S., Wilson, M. D., Vlasova, A., Qu, Z., & Zaucha, J. (2016). Study on DNA variants in Niemann-Pick disease. Journal of Genetic Research, 45(3), 215-230.
|
| [2] |
Garver, W. S., Jelinek, D., Meaney, F. J., Flynn, J., Pettit, K. M., Shepherd, G., & Heidenreich, R. A. (2010). Niemann-Pick disease mutations and prevalence. Molecular Genetics and Metabolism, 99(1), 10-15.
|
| [3] |
Vanier, M. T., & Millat, G. (2003). Niemann-Pick disease type C. Clinical Genetics, 64(4), 269–281.
|
| [4] |
Wassif, C. A., Cross, J. L., Iben, J., Sanchez-Pulido, L., Cougnoux, A., Platt, F. M., & Porter, F. D. (2016). High frequency of adult-onset Niemann-Pick disease type C. Orphanet Journal of Rare Diseases, 11, 12.
|
| [5] |
Wijburg, F. A., Sedel, F., Pineda, M., Hendriksz, C. J., Fahey, M., Walterfang, M., & Patterson, M. C. (2012). Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology, 78(20), 1560-1567.
|
| [6] |
Reunert, J., Fobker, M., & Panigrahi, I. (2013). Phenotypic variability in NPC-associated mutations. Journal of Inherited Metabolic Disease, 36(1), 121-129.
|
| [7] |
Steinberg, S. J., Dodt, G., Raymond, G. V., Braverman, N. E., Moser, A. B., & Moser, H. W. (2006). PEX gene mutations and peroxisome biogenesis disorders. Human Mutation, 27(11), 1097-1106.
|
Cite This Article
-
APA Style
Bishayee, A., Basu, S., Roy, S., Ray, A. C. (2025). A Rare Case of Adult-onset Niemann-Pick Disease Type C with PEX6 Mutation: Novel Genetic Insights. International Journal of Clinical and Experimental Medical Sciences, 11(2), 18-22. https://doi.org/10.11648/j.ijcems.20251102.11
 Copy
|
Copy
|
 Download
Download
ACS Style
Bishayee, A.; Basu, S.; Roy, S.; Ray, A. C. A Rare Case of Adult-onset Niemann-Pick Disease Type C with PEX6 Mutation: Novel Genetic Insights. Int. J. Clin. Exp. Med. Sci. 2025, 11(2), 18-22. doi: 10.11648/j.ijcems.20251102.11
Copy
|
Download
AMA Style
Bishayee A, Basu S, Roy S, Ray AC. A Rare Case of Adult-onset Niemann-Pick Disease Type C with PEX6 Mutation: Novel Genetic Insights. Int J Clin Exp Med Sci. 2025;11(2):18-22. doi: 10.11648/j.ijcems.20251102.11
Copy
|
Download
-
@article{10.11648/j.ijcems.20251102.11,
author = {Aditya Bishayee and Sagar Basu and Soumesh Roy and Avas Chandra Ray},
title = {A Rare Case of Adult-onset Niemann-Pick Disease Type C with PEX6 Mutation: Novel Genetic Insights
},
journal = {International Journal of Clinical and Experimental Medical Sciences},
volume = {11},
number = {2},
pages = {18-22},
doi = {10.11648/j.ijcems.20251102.11},
url = {https://doi.org/10.11648/j.ijcems.20251102.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ijcems.20251102.11},
abstract = {Background: Niemann-Pick disease type C (NPC) is a rare autosomal recessive neurodegenerative disorder. Adult-onset presentations are particularly uncommon and often misdiagnosed. Here, we report a rare case of NPC type C associated with a novel mutation in NPC1 and an additional mutation in the PEX6 gene, which has not been previously documented. Methods: A detailed clinical evaluation, laboratory investigations, imaging studies, and whole-exome sequencing were conducted. Results: A 29-year-old male presented with neuropsychiatric symptoms and splenomegaly. Genetic testing identified two significant mutations: c.3591+3G>T in NPC1 and c.2256del in PEX6. Clinical features were consistent with NPC type C1; however, no phenotypic expression of PEX6 mutation was observed, its potential modifying effects cannot be excluded. Conclusions: This case highlights the diagnostic challenges and genetic complexity of NPC, emphasizing the importance of genetic testing in rare diseases. Further studies are required to explore the clinical significance of co-occurring mutations. A lack of access to specific NPC therapies and long-term follow-up data remains a challenge in such rare cases.},
year = {2025}
}
Copy
|
Download
-
TY - JOUR
T1 - A Rare Case of Adult-onset Niemann-Pick Disease Type C with PEX6 Mutation: Novel Genetic Insights
AU - Aditya Bishayee
AU - Sagar Basu
AU - Soumesh Roy
AU - Avas Chandra Ray
Y1 - 2025/07/19
PY - 2025
N1 - https://doi.org/10.11648/j.ijcems.20251102.11
DO - 10.11648/j.ijcems.20251102.11
T2 - International Journal of Clinical and Experimental Medical Sciences
JF - International Journal of Clinical and Experimental Medical Sciences
JO - International Journal of Clinical and Experimental Medical Sciences
SP - 18
EP - 22
PB - Science Publishing Group
SN - 2469-8032
UR - https://doi.org/10.11648/j.ijcems.20251102.11
AB - Background: Niemann-Pick disease type C (NPC) is a rare autosomal recessive neurodegenerative disorder. Adult-onset presentations are particularly uncommon and often misdiagnosed. Here, we report a rare case of NPC type C associated with a novel mutation in NPC1 and an additional mutation in the PEX6 gene, which has not been previously documented. Methods: A detailed clinical evaluation, laboratory investigations, imaging studies, and whole-exome sequencing were conducted. Results: A 29-year-old male presented with neuropsychiatric symptoms and splenomegaly. Genetic testing identified two significant mutations: c.3591+3G>T in NPC1 and c.2256del in PEX6. Clinical features were consistent with NPC type C1; however, no phenotypic expression of PEX6 mutation was observed, its potential modifying effects cannot be excluded. Conclusions: This case highlights the diagnostic challenges and genetic complexity of NPC, emphasizing the importance of genetic testing in rare diseases. Further studies are required to explore the clinical significance of co-occurring mutations. A lack of access to specific NPC therapies and long-term follow-up data remains a challenge in such rare cases.
VL - 11
IS - 2
ER -
Copy
|
Download